



2022基因治疗行业报告:从根治愈罕见病,克服技术及生产挑战

随着国产产品陆续进入临床阶段,基因治疗产业“弯道超车”的说法不再是一纸空谈。在基因编辑技术和递送技术的影响下,基因治疗内部分化出了几个不同的赛道,其应用场景也不再受限于单基因遗传病。在这个产业快速迭代的过程中,如何抓住基因治疗产业的关键发展要素,摸清产业的发展逻辑,是策略制定中的关键点。
基于此,蛋壳研究院撰写了《2022基因治疗行业报告》,并将在报告中着重回答以下几个问题:
1. 基因治疗产业的投融资、市场规模发展趋势如何?
2. 关键技术点的发展成熟度如何?
3. 配套的CDMO产业发展节奏如何?是否已经足够支撑产业现阶段的发展需求?
4. 未来产业将如何解决技术、生产、商业化方面的挑战?
5. 目前基因治疗产业最值得关注的创新趋势是哪些?
为了弄清上述问题,蛋壳研究院在产业内进行了广泛的调研,并结合自己的研究内容,试图从行业概述、技术路径、发展机遇与挑战、未来趋势研判等维度全面解析基因治疗行业,以期为行业关注者及参与者提供有价值的行业信息。
(注:报告全文可在文末扫码下载)
基因治疗,从根治愈,前景广阔
1基因治疗,从根治愈,兼具临床优势&研发优势
基因治疗从根治愈,刚需强烈
基因治疗,从根治愈。基因治疗的核心在于精准打击了疾病根源——异常DNA,是一种根本性的治疗策略。基因治疗,通常指将正常的目标基因导入人体靶细胞,或将异常基因敲除的治疗方式。
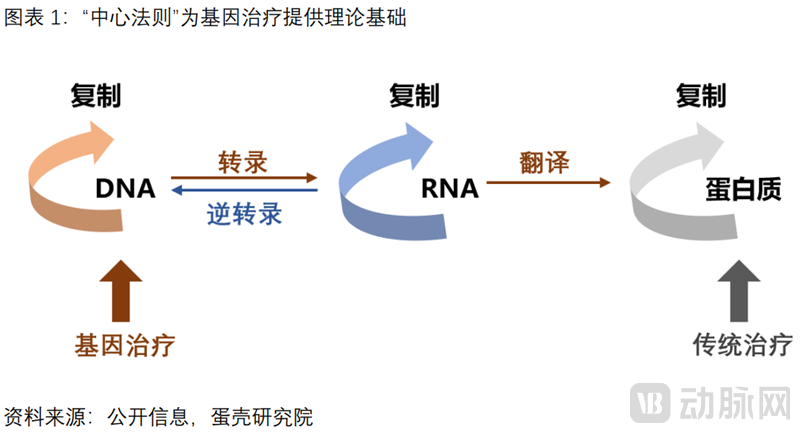
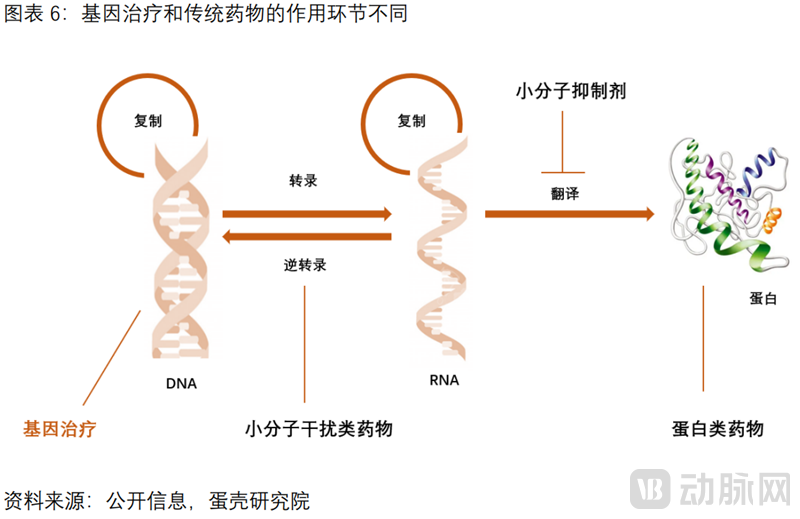
“中心法则”为基因治疗提供了理论基础。中心法则:遗传信息沿着“DNA-RNA-蛋白质”的路径传递,疾病发生时多表现为蛋白质层面的异常。传统治疗干预蛋白质,而基因治疗是从指导蛋白质合成的根源——DNA入手,通过调控DNA来改变遗传信息传递,从而改变蛋白质的性状,实现从根源上治疗疾病。
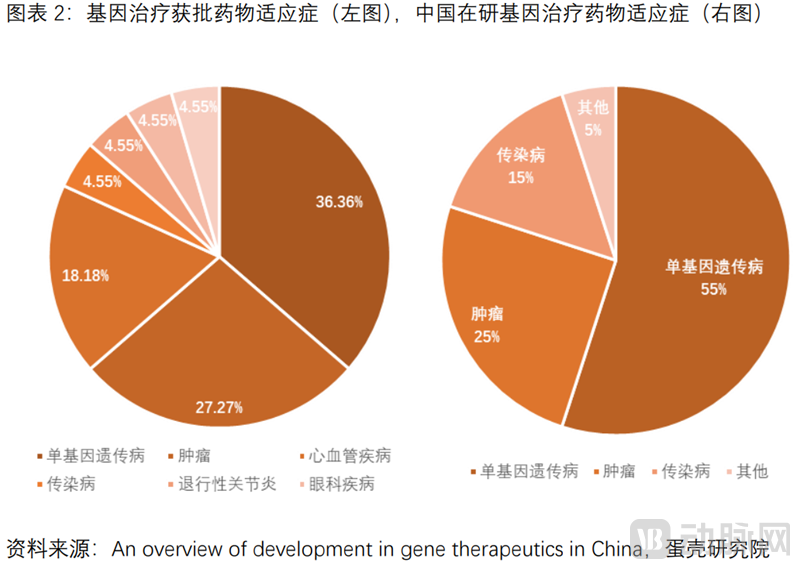
基因治疗刚需强烈,适应症以单基因遗传病(罕见病)为主,这类疾病的致病基因明确,同时缺乏有效的治疗手段,面临巨大的未满足临床需求。绝大多数的罕见病由基因异常导致,种类多达7000余种,总人数达3.5亿。然而,超90%罕见病缺乏有效的治疗手段。适应症以罕见病为主,包括眼科遗传病、血友病、地中海贫血、运动神经元疾病等。
基因治疗包括基因增补和基因编辑两种技术路径
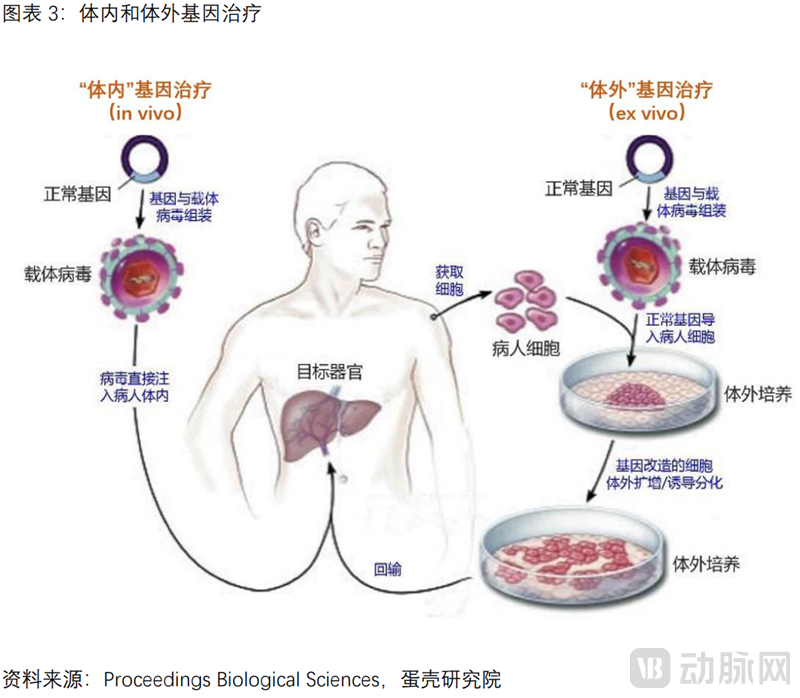
根据治疗途径,可将基因治疗划分为2类:体内基因治疗和体外基因治疗。“体内”基因治疗,操作流程相对简单,将携带治疗性基因的重组载体直接递送到患者体内。“体外”基因治疗,额外涉及细胞层面(自体造血干细胞)的体外遗传修饰,包括细胞的分离&转染&扩增培养&回输等。
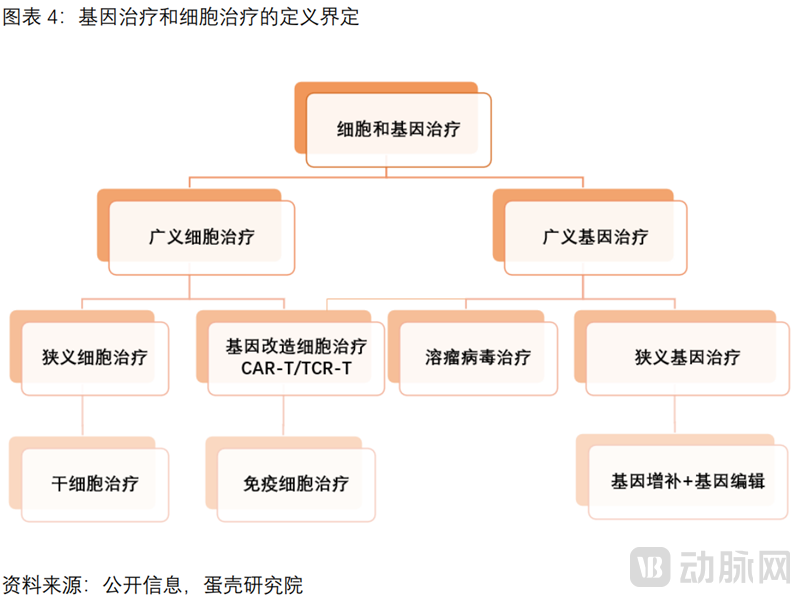
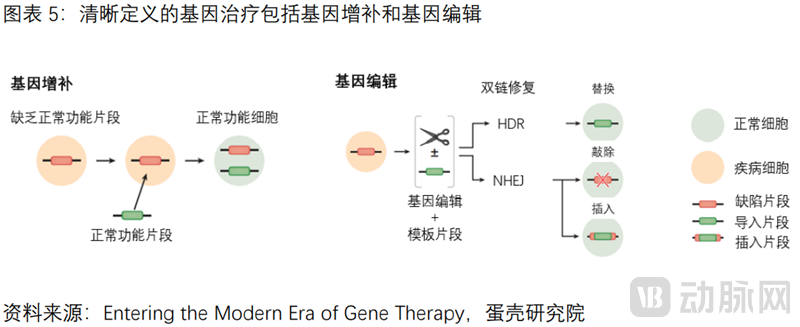
本报告清晰定义的基因治疗,是基于DNA层面进行的干预。主要包括基因增补、基因编辑两大技术路径;剔除:CAR-T等免疫细胞疗法,溶瘤病毒,小核酸药等。
(1)基因增补:利用递送载体,将外源基因导入病变细胞,其表达产物能修饰缺陷细胞的功能或加强原有功能。基因增补是目前获批上市和临床在研阶段产品中,最主要的基因疗法技术路径。
(2)基因编辑:精确修饰特定目标基因,从而破坏有害基因或修复变异基因,包括ZFNs,TALEN以及2020年获诺贝尔化学奖的CRISPR/Cas9技术。以CRISPR/Cas9技术为例,Cas9蛋白在sgRNA的导向下,通过碱基互补配对,到达不同的靶部位,通过切割靶基因,对目标基因进行定点精确编辑,从而实现对患者原有基因组“错误”基因的改变与修正。基因编辑系统向临床的转化正处于早期阶段,目前尚无产品上市。
相比于传统药物,基因治疗兼具临床优势及研发优势
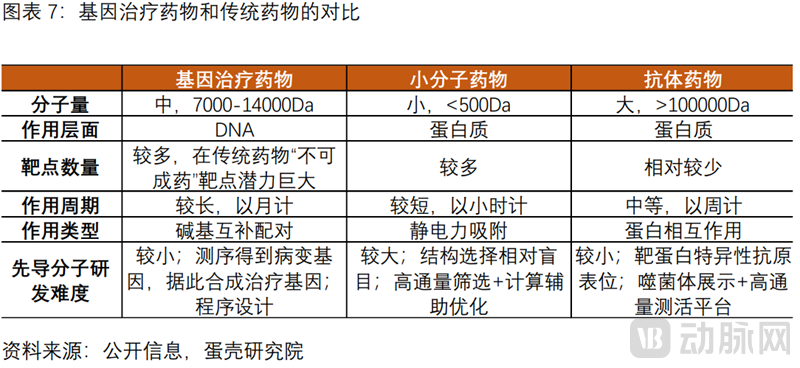
基因治疗的临床优势,体现在DNA层面直接干预治疗,避免传统药物在蛋白质层面“不可成药”靶点的困境。目前大部分药物以蛋白质为靶点,如治疗肿瘤的小分子靶向药和大分子单抗。基因治疗直接在DNA层面修正致病基因,绕过传统药物在成药性上的难点,对致病基因清晰而蛋白质水平难以成药的疾病具有独特的临床优势。
基因治疗的研发优势,体现在核酸序列的合成难度较传统药物更低。基因治疗的三大共性步骤:核酸序列的设计与合成、目标序列递送至靶细胞和工业化生产。其中,核酸序列的设计与合成难度较小分子靶向药和单抗更低,因此一旦研发出一个安全高效的递送方式,基因治疗产品的研发难度反而更低。
2曲折中前行,基因治疗未来已来
经蛋壳研究院汇总梳理,基因治疗的发展可分为初期探索、狂热发展、曲折前行、再度繁荣4个阶段,下面列示了各个时期的标志性事件。
1963年,美国分子生物学家、诺贝尔生理学或医学奖获得者JoshuaLederberg首次提出“基因交换和基因优化”概念,标志着基因治疗的起点。
1970年,美国医生StanfieldRogers试图通过注射含有精氨酸酶的乳头瘤病毒来治疗一对姐妹的精氨酸血症,这是首例人体试验,试验以失败告终。
1990年,“基因治疗之父”WilliamFrenchAnderson医生领衔开展了全球首例针对重症联合免疫缺陷病的基因治疗,患者为一名美国4岁女孩。接受治疗后,其机体产生腺苷脱氨酶的能力有所提高,病情得到缓解,该患者目前仍然存活。
1996年,ZFN基因编辑技术发明。
1999年,美国男孩JesseGelsinger参与了宾夕法尼亚大学的基因治疗项目,接受治疗4天后因病毒引起的强烈免疫反应导致多器官衰竭而死亡。该事件是基因治疗发展的转折点。
2003年,FDA暂时中止了所有用逆转录病毒来改造血液干细胞基因的临床试验,但经过3个月严格审核权衡后,又允许基因治疗临床试验继续进行。
2011年,TALEN基因编辑技术发明。
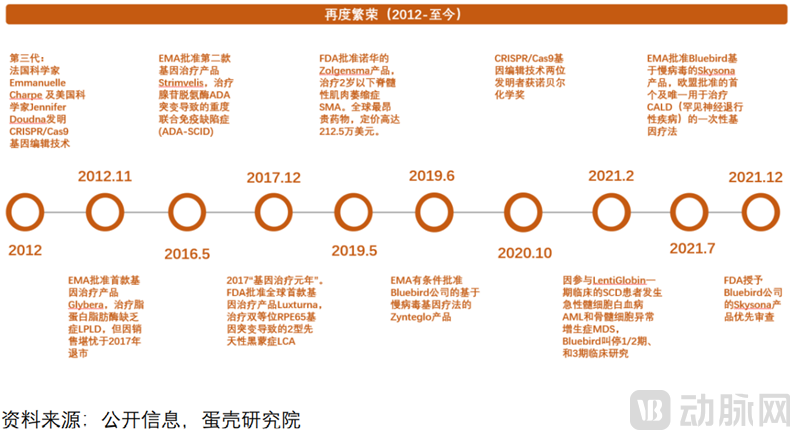
2012年,美国科学家JenniferDoudna 及法国科学家EmmanuelleCharpentier发明了CRISPR/Cas9基因编辑技术,这是基因治疗领域革命性的事件。
2017年12月,FDA批准了全球首款基因治疗产品Luxturna,用于治疗双等位RPE65基因突变导致的2型先天性黑蒙症LCA。2017年被称为基因治疗“元年”。
2020年10月,CRISPR/Cas9基因编辑技术发明者获得诺贝尔化学奖。法籍微生物学家EmmanuelleCharpentier博士和美国国家科学院院士JenniferA.Doudna博士获得了2020年诺贝尔化学奖,这两位女科学家共同发现了Cas9的切割作用和crRNA的定位作用,并将crRNA与tracrRNA可以融合成单链引导RNA(sgRNA)。
3政策助力,资本狂热,基因治疗前景广阔
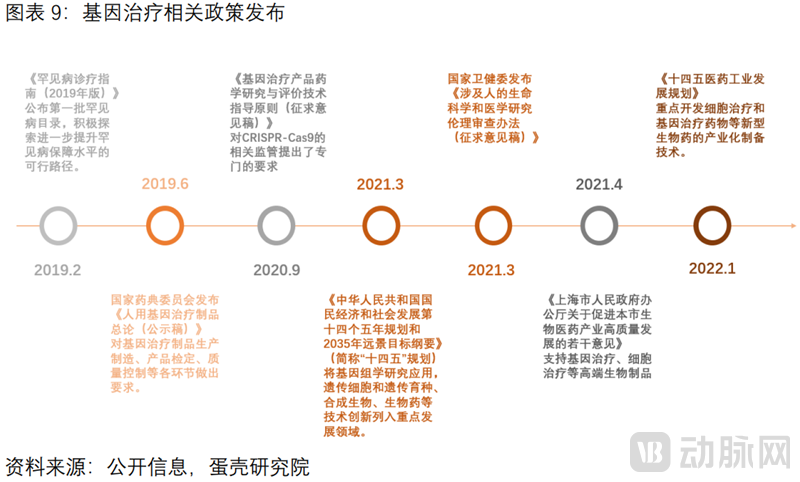
政策助力基因治疗领域健康发展。无论是“十四五”规划将基因组学研究纳入重点发展领域,罕见病相关政策红利推动疾病诊疗,还是监管政策对CRISPR-Cas9等基因编辑工具的具体规范要求,都让基因治疗领域的发展更有迹可循。
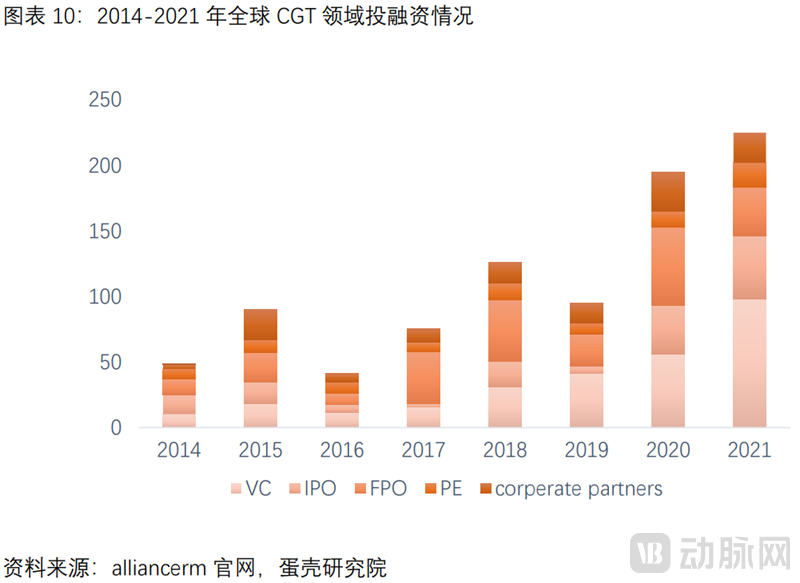
全球细胞与基因治疗投融资火热。随着2017年FDA批准Luxturna,Kymriah和Yescarta以来,CGT行业的快速发展吸引了大量资本的流入,据alliancerm披露,全球CGT领域投融资总额从2014年约50亿美元快速增长至2021年约230亿美金。
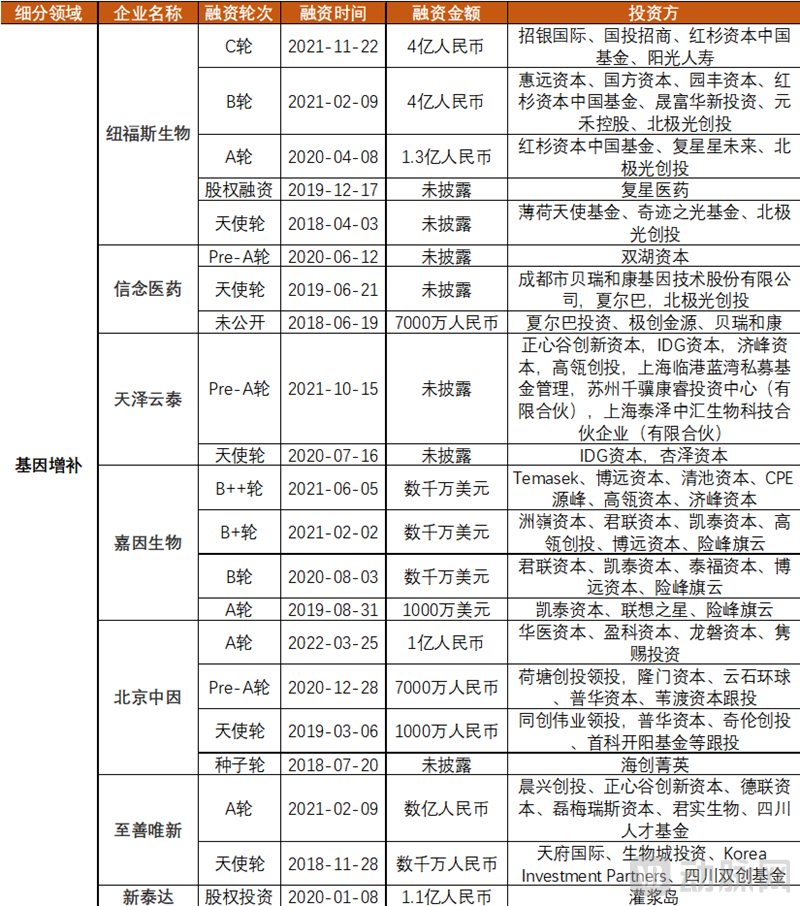
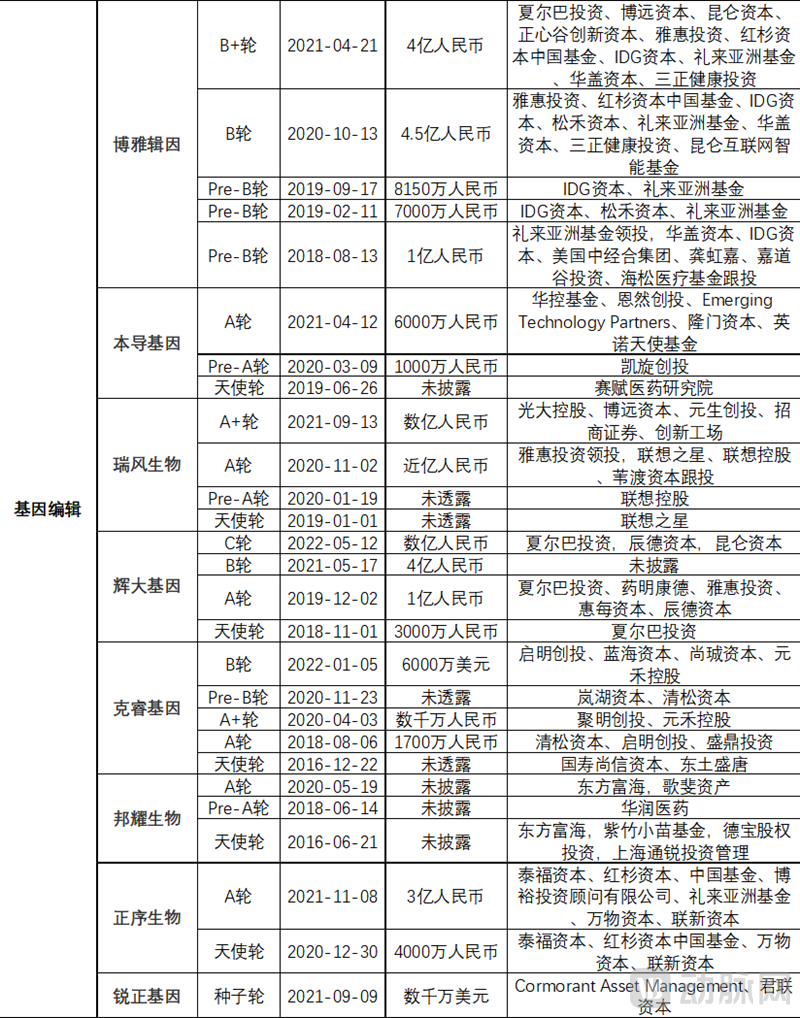
国内基因治疗领域多家企业频获融资,助推创新发展。无论是基因 治疗药物研发的基因增补还是基因编辑领域,近年多家企业获得大额融资。例如纽福斯生物,杭州嘉因生物,至善唯新,博雅辑因,瑞风生物,辉大基因,正序生物,锐正基因等,单轮即获数亿元融资。同时,近年为CGT提供CDMO服务的企业也融资频繁,例如2022年3月22日科创板上市的和元生物,以及金斯瑞,宜明细胞,派真生物等,均获单轮数亿元融资。
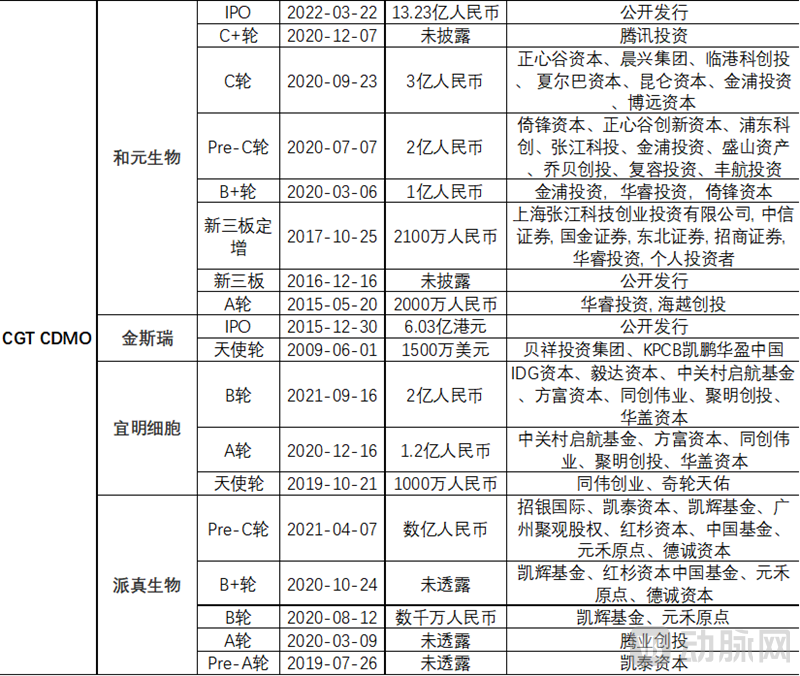
资料来源:动脉橙数据库,公开信息,蛋壳研究院
基因治疗飞速发展,前景广阔。预计2025年全球及中国规模达305.4亿美元和178.9亿元,2020-2025年CAGR高达71.2%和276.0%。
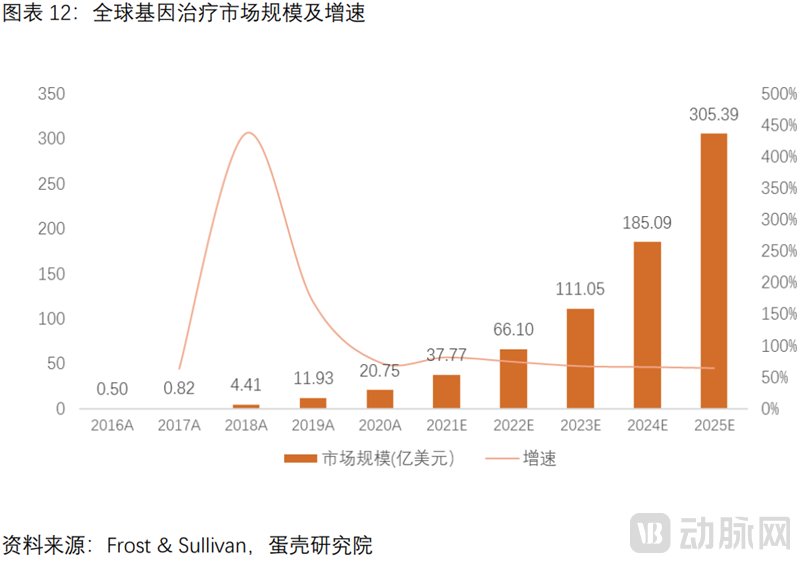
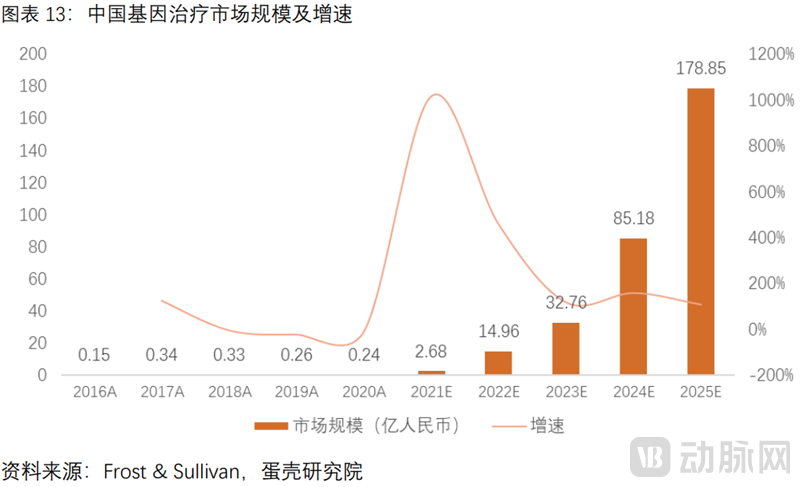
基因治疗有效治愈罕见病,病毒载体是基因治疗的关键钥匙
1两大技术路径:基因增补相对成熟,基因编辑“功能强大”+“定向精准”
基因增补技术相对成熟,已有数款上市产品
基因增补技术相对成熟。2012年至2021年,FDA批准了2款产品,EMA额外批准了4款,适应症集中于罕见病领域。
FDA批准了2款产品,均为基于AAV载体的体内疗法。2017年批准Spark的Luxturna,治疗双等位RPE65基因突变导致的2型先天性黑蒙症LCA,以及2019年批准的Zolgensma,治疗2岁以下的脊髓性肌肉萎缩症SMA。
EMA额外批准了4款产品。2012年批准uniQure的Glybera,基于AAV载体,是EMA批准的首款产品,治疗脂蛋白脂肪酶缺乏症LPLD,但由于销售堪忧,被迫于2017年10月退市。2016年,EMA批准了第二款产品,GSK的Strimvelis,基于RV载体,治疗腺苷脱氨酶ADA突变导致的重度联合免疫缺陷症(ADA-SCID),2018年4月,GSK出售给Orchard,仅5例患者接受治疗。2019年和2021年,EMA先后批准了Bluebird基于LV载体的Zynteglo和Skysona,分别治疗12岁及以上的非β0/β0基因型输血依赖性β-地中海贫血(TDT),和早期脑性肾上腺脑白质营养不良(CALD)。
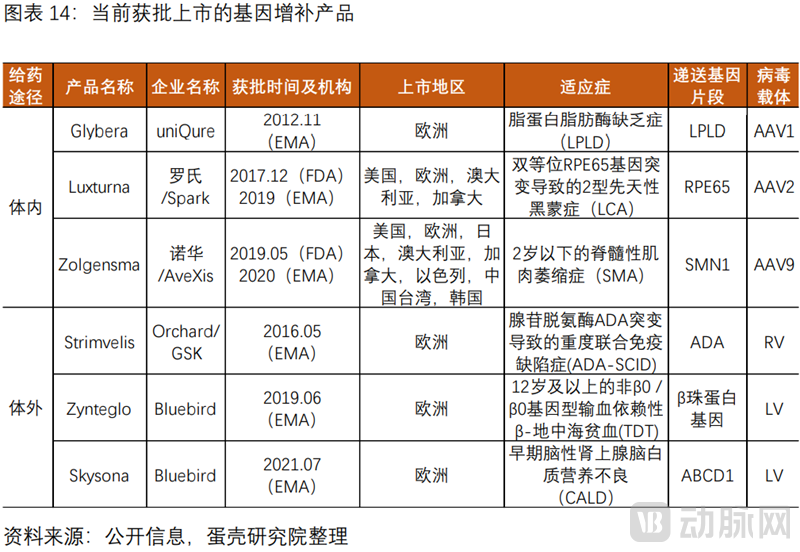
(1)基因增补在研管线丰富,海外不少产品已进入拟上市/上市申请或临床后期阶段
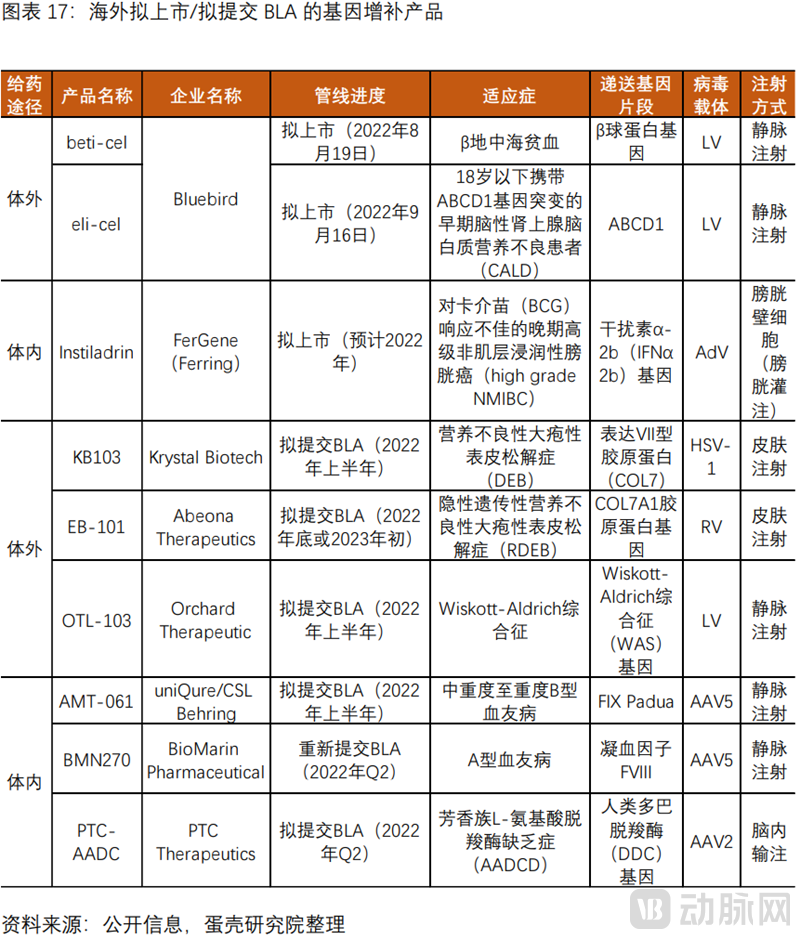
海外基因增补产品的在研管线丰富。经蛋壳研究院汇总整理,2022年即将有3款拟上市产品,以及6款拟提交上市申请BLA的产品,以及十几款已经进入临床3期的在研管线。纵观海外基因增补技术的在研管线,我们发现,产品以体内基因治疗为主,且基于AAV载体;体外治疗大多基于LV载体,包括Bluebird公司的两款拟上市产品beti-cel和eli-cel,PDUFA目标日期分别为2022年8月19日和2022年9月16日。不少产品目前beti-cel疗法已获FDA优先审评资格,如果获得FDA的批准,预计该疗法会成为潜在的美国首个针对β地中海贫血患者的慢病毒载体基因疗法。此前FDA曾授予Instiladrin产品快速通道资格,突破性疗法认定和优先审评资格,并接收递交的BLA。如果2022年获得批准,该疗法将为对BCG无反应的NMIBC患者提供一个有希望的选择。OTL-103产品已获得FDA授予的孤儿药资格和罕见儿科疾病(RPD)资格。Etranacogenedezaparvovec产品可能是潜在的第一个为血友病B患者提供持久、功能性治疗益处的基因疗法。BMN270产品获得了FDA和EMA的孤儿药指定,用于治疗重度血友病A。
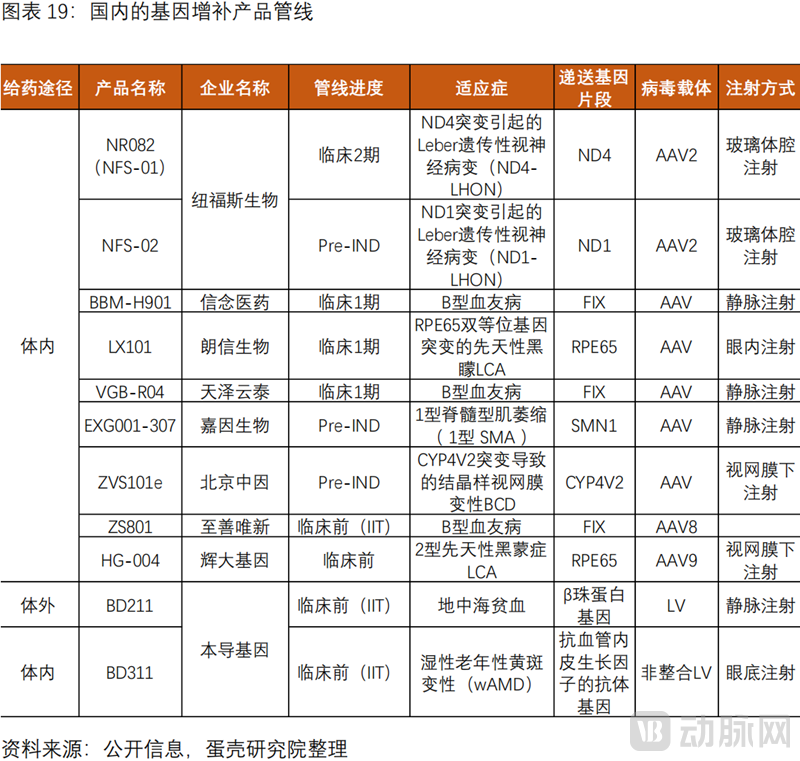
(2)国内基因增补管线整体进展较慢,适应症集中于眼科遗传病及血友病
国内基因增补管线整体进展较慢,多处于临床1期,适应症集中于眼科遗传病和血友病。进展较快的管线包括纽福斯生物的NR082(NFS-01),信念医药的BBM-H901,朗信生物的LX101,天泽云泰的VGB-R04,杭州嘉因生物的EXG001-307,以及北京中因的ZVS101e等。
纵观国内基因增补技术的在研管线,我们发现,产品基本为体内途径,且基于AAV载体,仅本导基因基于LV平台有相关管线。数家国内研发药企的管线获得FDA的孤儿药认定(ODD),包括纽福斯生物的NR082(NFS-01),天泽云泰的VGB-R04,北京中因的ZVS101e。
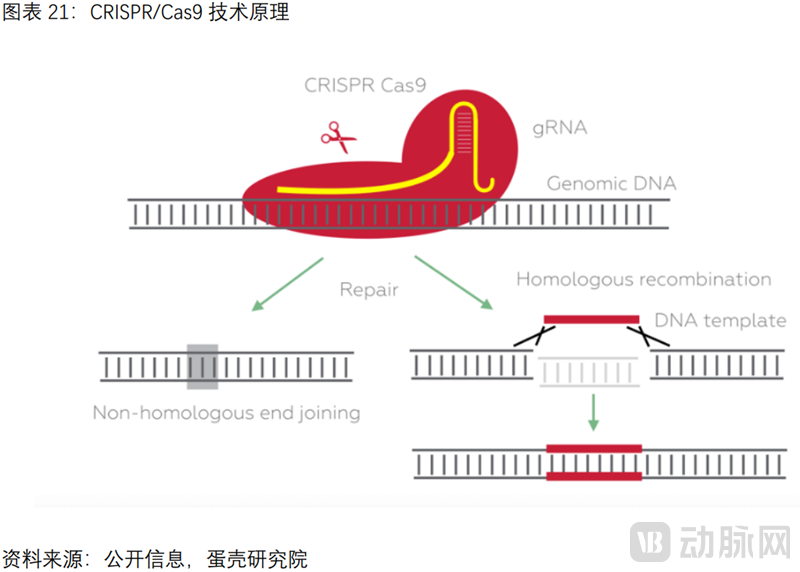
基因编辑技术定向精准,功能强大
基因编辑“功能强大”+“定向精准”,2020年获“诺奖”的CRISPR引领革命性突破。
“功能强大”:基因编辑可实现“基因敲除”(非同源末端连接NHEJ)和“基因插入”(同源重组HRD)。而基因增补只能介导“基因插入”。“定向精准”:CRISPR/Cas9系统通过sgRNA的碱基互补配对精准到达靶基因。而基因增补通常采用AAV或LV递送,AAV不整合宿主基因组,面临耐久性问题;LV随机插入基因组,面临致癌风险。
CRISPR/Cas9的核心优势在定位方式,sgRNA的合成较蛋白容易,因此具有高效便捷、成本低廉的优势。CRISPR/Cas9是目前应用最广泛的基因编辑工具,基于细菌获得性免疫系统,防御病毒入侵的机制。2020年10月,CRISPR/CAS9基因编辑发明者获诺贝尔化学奖。
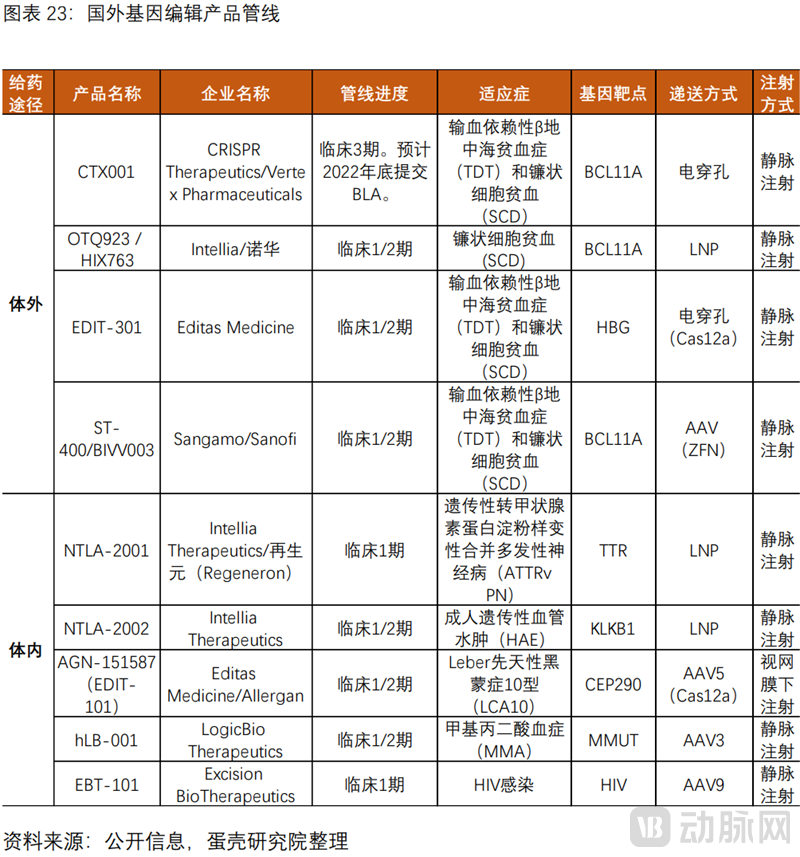
(1)海外基因编辑管线基本由“三巨头”包揽
基因编辑的海外管线基本由“三巨头”包揽,进展最快CRISPR的CTX001预计2022年底提交BLA。在研管线集中于CRISPR/Cas9,三位发明者Emmanuelle Charpentier,Jennifer Doudna,以及张锋教授,分别创立了全球领先的基因编辑公司——CRISPR,Intellia,Editas。国内基因编辑管线整体仍处于临床早期阶段。
CRISPR的CTX001是全球进展最快的体外编辑疗法。计划2022年底提交BLA,治疗输血依赖性β地中海贫血症(TDT)和镰状细胞贫血(SCD),已获得FDA授予再生医学高级治疗产品(RMA)、快速通道资格(FTD)和孤儿药资格(ODD),欧盟委员会ODD。最新进展:截至2021年3月30日,所有15名TDT患者均不依赖输血,所有7名SCD患者均未出现血管闭塞危象(VOCs)。
Intellia的NTLA-2001是创新性的体内编辑疗法。采用LNP递送全核酸基因编辑药物(cas9mRNA+sgRNA),用于治疗遗传性转甲状腺素蛋白淀粉样变性合并多发性神经病(ATTRvPN)。2021年10月,FDA授予孤儿药资格(ODD)。最新进展:截至2022年3月1日,15名患者的血清TTR水平表现出依赖性降低,且无严重不良事件。
Editas的AGN-151587(EDIT-101)是全球首个体内编辑疗法。治疗Leber先天性黑蒙症10型(LCA10)。早期临床结果:仅证实了安全性,在疾病改善方面并不理想。
(2)国内基因编辑管线整体仍处于临床早期阶段
国内基因编辑管线整体仍处于临床早期阶段。进展最快的是博雅辑因的体外疗法ET-01,处于临床1期,体外疗法还有邦耀生物的BRL101,瑞风生物的RM001。体内疗法包括本导基因的BD111,北京中因的ZVS203e,辉大基因的HG-203,以及锐正基因的相关管线。
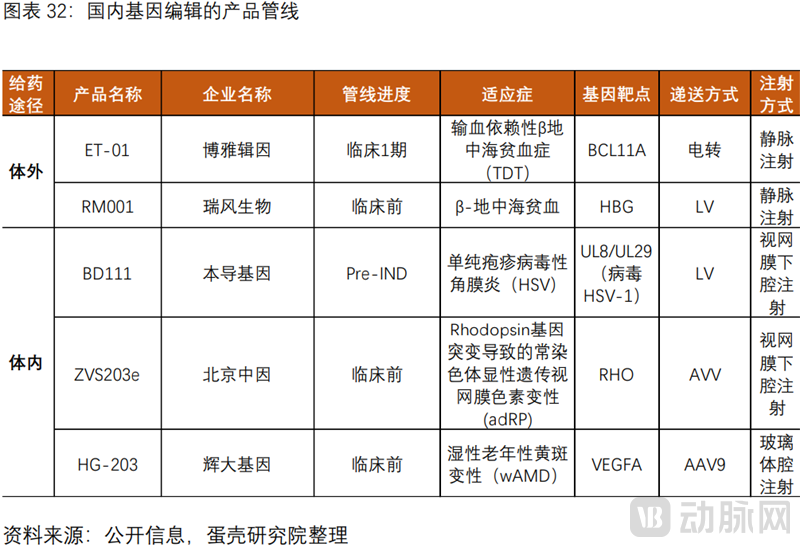
2病毒载体是基因治疗的关键钥匙,其中AAV以安全性优势最为广泛使用
基因治疗的一大核心是递送方式,理想的递送方式需具备多项要素。首先能够剔除复制自身载体的能力,具有高转导效率;能靶向特定的细胞,且可以长期稳定表达转基因;具有较低的免疫原性,不会引起炎症。
目前被广泛使用的递送方式:经人工改造失去致病能力的病毒载体,其关键优势在于天然转导效率高。据ASGCT,89%在研CGT项目采用病毒载体进行递送。
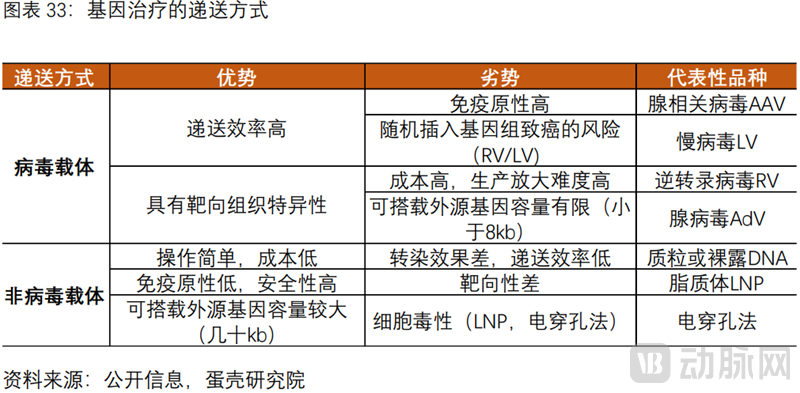
常见的病毒载体:逆转录病毒载体RV,慢病毒载体LV,腺相关病毒载体AVV和腺病毒载体AdV。
AAV被广泛应用的核心优势:目前最安全的病毒载体,免疫原性低,非致病性;不整合宿主基因组避免致癌风险,天然AAV血清型提供组织靶向特异性。
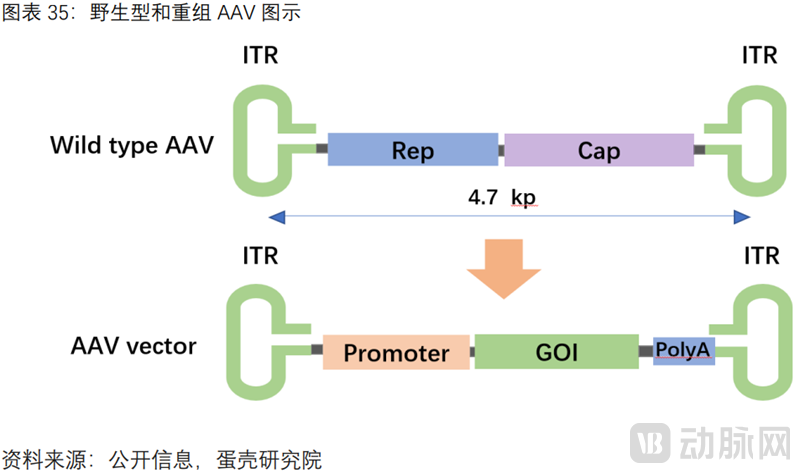
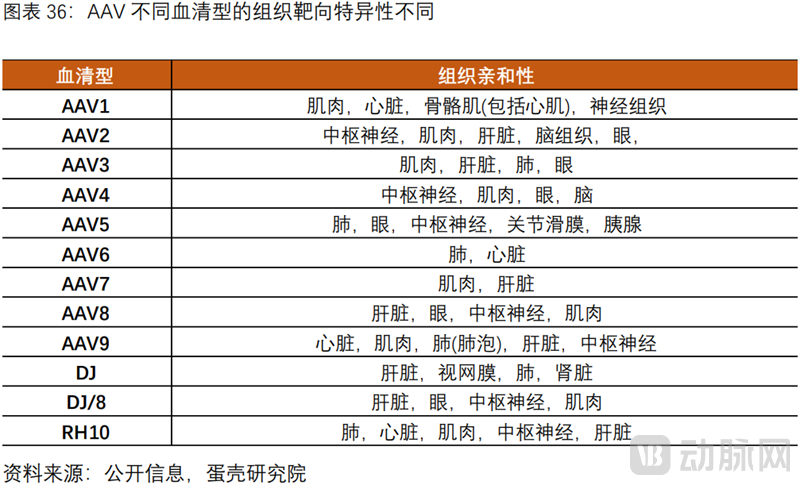
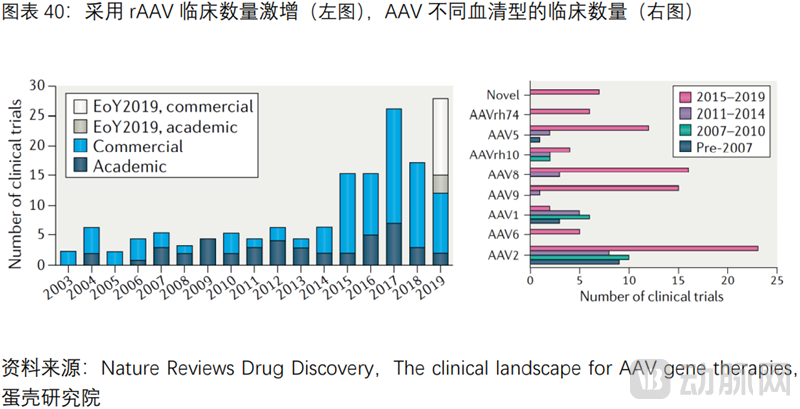
CGT CDMO解决病毒载体规模化生产的瓶颈,助推商业化进程
1基因治疗产业链的上游主导病毒载体的生产,是基因治疗商业化的核心
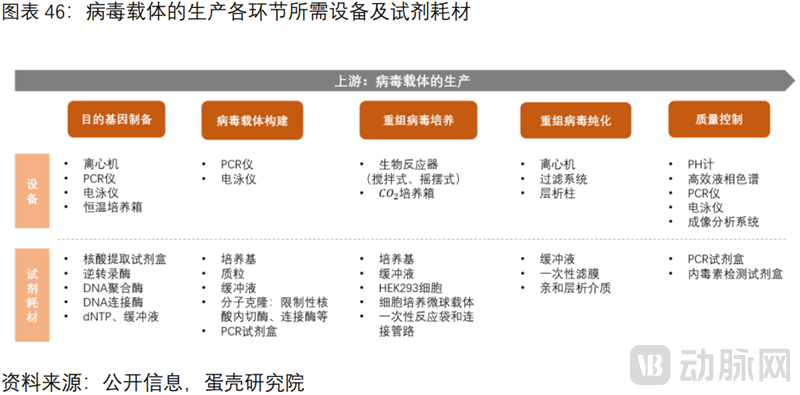
基因治疗产业链可分为上游,中游和下游。上游是病毒载体的生产厂商。病毒载体的生产步骤包括:目的基因制备,病毒载体构建,重组病毒培养,重组病毒纯化,质量控制等环节,各环节涉及多种设备及试剂耗材。中游是基因治疗的制药企业,包括基因增补,基因编辑两类药企。下游则是各类罕见病/遗传病患者。包括2型先天性黑蒙症(LCA),脊髓性肌肉萎缩症(SMA),β-地中海贫血(TDT),A/B型血友病,遗传性视网膜病变等。

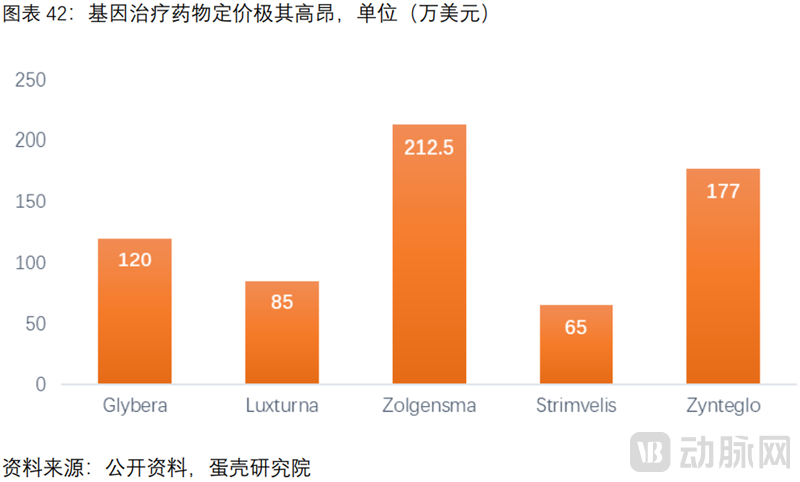
基因治疗产品的商业化定价普遍极其高昂。“全球最昂贵药物”Zolgensma,定价高达212.5万美元(合人民币1400万元)。其余几款产品的平均治疗费用也在100万美元以上。
高昂的定价阻碍商业化进程。EMA于2012年和2016年批准的两款基因治疗产品Glybera和Strimvelis,均因商业化销售堪忧,分别于2017年被迫退市和2018年被GSK出售。
控制病毒载体的生产成本是终端产品合理定价的关键。
(1)病毒载体占1/3成本:据《纽约时报》报道,基因治疗研发费用中1/3用于上游病毒载体的生产制备,降低病毒载体的生产成本能有效控制终端定价。
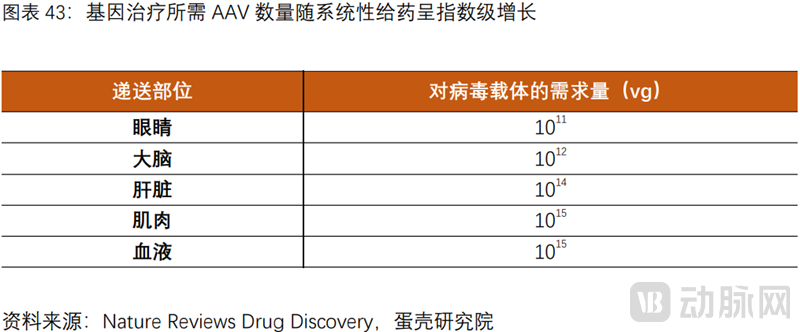
(2)给药范围的扩大对载体需求量呈指数级增长,带来终端定价的显著攀升:Luxturna视网膜下腔注射,定价85万美元,而全身静脉注射的Zolgensma售价高达212万美元。因为从局部的眼部注射,到大脑,再到肌肉,甚至全身血液注射,每个患者所需的病毒载体数量呈指数级增长。
2病毒载体的规模化生产存在诸多壁垒,产能极度短缺
病毒载体的生产涉及多项工艺,步骤繁琐。上游:病毒构建和细胞扩增培养,下游:细胞裂解、澄清、浓缩、层析纯化、精制等多项工艺。
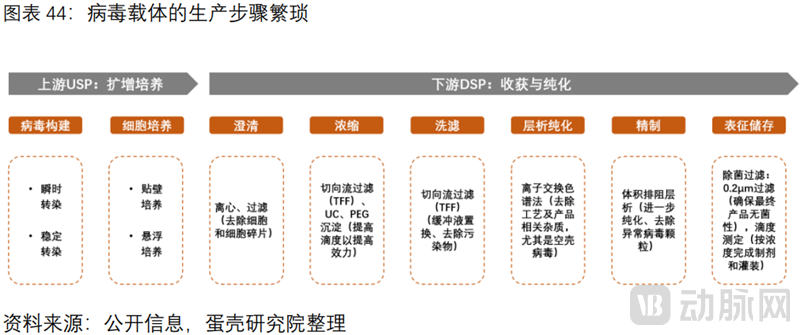
(1)病毒载体的生产面临诸多工艺壁垒及人才壁垒。上游难点:如何减少瞬时转染所需的质粒数量;如何扩大细胞培养的产能等。下游难点:层析纯化环节,DSP整体收率仅20-30%,难点在于USP中存在的空壳病毒。国内具备病毒经验、工艺背景和生产管理经验的复合型人才极度稀缺。
(2)病毒载体的生产面临很高的资金壁垒。为建立符合cGMP标准的厂房及设备,需重资产投入(数亿美元),且各环节需要多种设备及试剂耗材,基本依靠进口。设备包括生物反应器,离心机,层析柱等,试剂耗材包括质粒、培养基、HEK293细胞等。
全球病毒载体的产能极度短缺,且短期无法解决产能瓶颈。据L.E.K.统计,CGT CDMO全球平均等待时间长达16个月,甚至2年。据RootsAnalysis分析,当前产能缺口至少在1-2个数量级,未来才能满足产品需求。
3CGT CDMO解决病毒载体的生产瓶颈,产业链上必不可少的参与者
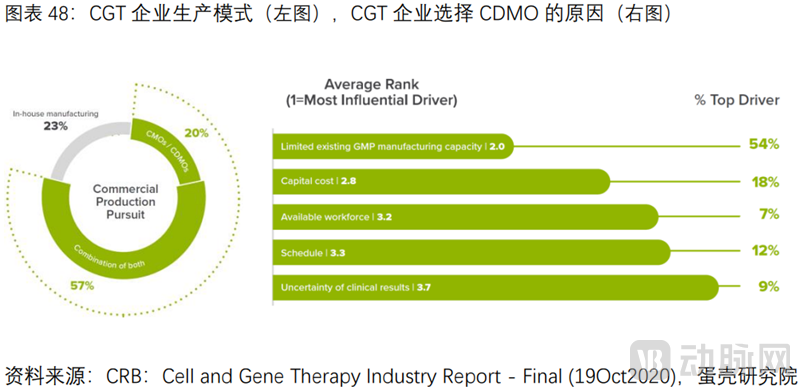
CGT CDMO降本增效,生产外包渗透率65%远高于传统药物35%。2021年,CRB对150家企业调研显示,仅23%选择完全自主搭建产线,绝大多数企业选择完全(20%)或部分(57%)外包。
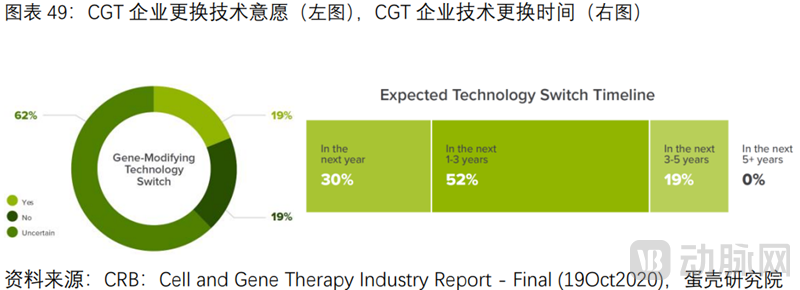
CDMO是产业链上必不可少的参与者。调研显示,企业选择外包的主要原因:缺乏GMP级生产产能(占54%),18%和12%企业出于研发成本和周期考虑。另外,高达81%公司未来5年可能更换研发技术,而FDA要求IND时必须明确生产工艺,一旦有重大变更需重新申报。因此CDMO为药企提供了多样化工艺的灵活选择。
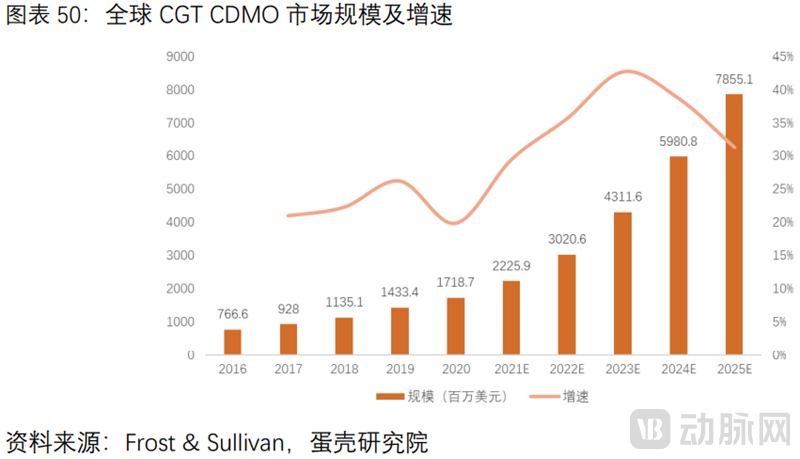
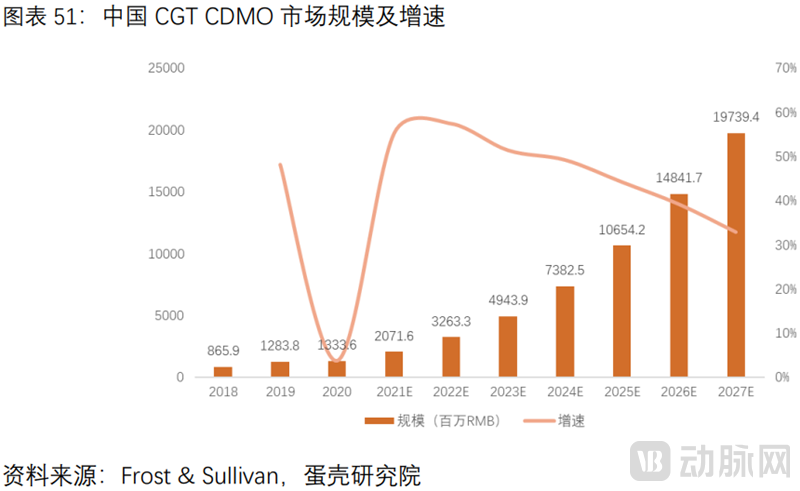
CGT CDMO市场快速扩容。据沙利文,全球CGT CDMO市场规模从2016年7.67亿美元增长至2020年17.19亿美元,年复合增长率22.4%。预计2025年市场规模达78.66亿美元,2020年至2025年复合增长率达35.5%。中国CGT CDMO从2018年至2022年,市场规模由8.7亿人民币预计增至32.6亿人民币,预计2027年市场规模达197.4亿人民币。CGT CDMO将迎来黄金期,未来将有更多公司通过并购或扩张布局这一领域。
三维度剖析基因治疗的挑战及趋势展望(技术+生产+商业化)
1基因治疗面临技术&生产&商业化的多重挑战
(1)技术挑战
由于基因增补技术中的目的基因合成较容易,因此增补技术的难点落在递送载体本身,其技术挑战包括转导效率(目标基因到达靶细胞的效率)、靶向组织特异性(脱靶细胞中的基因表达可能导致毒性或触发不必要的免疫反应)、AAV载体容量(仅4.7kb,无法搭载过大片段)以及免疫障碍(主要包括适应性免疫反应如CTL应答、中和抗体NAbs,以及对AAV转导后的先天免疫反应)。
基因编辑最关键的技术挑战是脱靶效应引发的安全性问题。野生型Cas9蛋白切割双链DNA形成双链断裂(DSBs),从而增加脱靶效应的几率。2022年3月2日,Nature期刊研究显示,当sgRNA第18-20个碱基错配时,Cas9酶并没有放弃前进,而是通过一个手指状结构紧紧抓住错配区,从而稳定了RNA-DNA双链,使其表现地像正确配对,为Cas9对DNA的切割铺平道路,从而导致严重的安全性问题“脱靶效应”——即CRISPR编辑系统对体内非目标区域的DNA双链进行了错误切割。
(2)生产瓶颈
病毒载体生产的上游(USP)难点包括:如何减少瞬时转染所需的质粒数量以及提高转染效率,从而降低质粒成本(AAV主要成本来源);如何提高细胞培养密度,扩大产能。
下游(DSP)主要难点在层析纯化环节,尤其是去除空壳病毒。因为AAV常用于体内基因治疗,对纯度要求高。但产品相关杂质如空壳病毒、降解产物等较难去除。目前病毒载体DSP整体收率仅20-30%,难点在于上游USP粗液中大量存在的空壳病毒(占比高于70%):(1)不含有治疗基因但衣壳蛋白本身会引起免疫原性风险(2)和完整病毒竞争感染细胞表面有限的受体,导致转导效率降低。
(3)商业化困境
一方面,罕见病适应症的患者基数本身较少。Glybera用于治疗脂蛋白脂肪酶缺乏症LPLD,该疾病过于罕见,发病率约为1/100万且误诊率较高。上市期间仅1位患者接受了该治疗。Strimvelis用于治疗腺苷脱氨酶ADA突变导致的重度联合免疫缺陷症(ADA-SCID),该疾病极为罕见,每年欧洲仅新增15例患者,因此截至2017年,仅2名患者接受治疗。直到2018年4月,GSK将Strimvelis出售给Orchard时,仅5例患者接受了该治疗。
另一方面,基因治疗产品的商业化定价极其高昂。Zolgensma定价高达212.5万美元(合人民币1400万元)/次,有“全球最昂贵药物”之称,其余几款产品的平均治疗费用也在100万美元以上。基因治疗研发成本过高导致的终端商业化定价极其高昂,加之保险支付体系欠缺,导致实际有支付能力的患者寥寥无几。
本文来自其他网站,不代表健我头条立场和观点,如若转载,请注明出处:https://news.jianwo.com/detail_39838.html如有版权问题,请联系客服配合您删除这篇文章。
-
0

-

参与讨论

动脉网
最近文章
相关文章
- 2022-06-21
对话以色列VirtuOptica:VR游戏自动验光配镜产品,打破百年传统配镜模式
2022-06-21微脉、动脉网、DTA联合发布《数字疗法价值评估和整合指南》,解码数字疗法应用
2022-06-17易参正式发布《2022 年度科创板生物医药行业股权激励报告》
2022-06-17
关注我们








